Abstract
Introduction: Constitutional Mismatch Repair Deficiency (CMMR-D) syndrome is a rare tumour predisposition and polyposis syndrome that presents in childhood. It is caused by mutations in mismatch repair (MMR) genes that result in a tumour spectrum including colorectal cancers, high-grade gliomas, non-Hodgkin T-cell lymphomas and leukaemias. It is characterized by biallelic germline mutation of one of four MMR genes that can be identified by immunohistochemistry. Immunohistochemistry is used in screening for Lynch syndrome (LS); however, the pattern of loss-of-staining in the background, non-tumour tissue is unique to CMMR-D syndrome. CMMR-D syndrome is seen in LS families and occurs as a result of consanguinity or founder effect. In South Africa, LS families in the Western and Northern Cape Provinces show a unique MLH1 c1528C>T mutation. The diagnosis of CMMR-D syndrome includes clinical findings outlined in the European Consortium’s Care of CMMRD document and confirmation of biallelic mutation in one MMR gene. Immunohistochemistry can be used in the diagnosis of CMMR-D syndrome by identifying cases for targeted molecular genetic tests. Loss of staining of the affected gene in the background, non-tumour tissue, is a key feature of CMMR-D syndrome. Methods: A retrospective analysis of archival, formalin fixed paraffin embedded tissue was performed on specimens of children attending Red Cross Children’s Hospital with tumours that form part of the CMMR-D spectrum, outlined by the Care for CMMRD criteria. We used the criteria of high-grade gliomas (WHO Grade III or IV) occurring before 25 years of age, cutaneous lesions suggestive of CMMR-D syndrome and patients with a first or second degree relative diagnosed with LS. Immunohistochemistry was performed and the staining pattern was documented using a modified Allred Scoring system. Specific attention was given to the characterization of the staining pattern of the background normal tissue. Results: 21 samples evaluated from 18 patients. 16 samples represented brain tumours. Three inadequate samples were excluded. 12 samples showed intact staining. Two samples showed staining of unknown significance. Four samples from 3 different patients showed staining patterns compatible with MMR deficiency. Of these four samples, three samples showed loss of staining in background non-tumour tissue with positive external control. Conclusion: MMR immunohistochemistry can be used in the evaluation of CMMR-D syndrome. The pattern and scoring of both the tumour and the background non-tumour tissue is critical. The diagnosis of CMMR-D syndrome depends on clinical application of Care for CMMRD criteria, MMR immunohistochemistry in conjunction with molecular genetic testing.
Keywords
Pediatric, Mismatch Repair Deficiency Syndrome, Glioma, Histology, Immunohistochemistry, Tumour Syndrome, Oncology
1. Introduction
The Mismatch Repair (MMR) system is responsible for the detection and repair of DNA base pair mismatches during DNA replication in human prokaryotic and eukaryotic cells. The MMR system is also involved in the apoptotic response
. Four Mismatch Repair (MMR) genes encode four corresponding MMR proteins:
MSH2,
MSH6,
MLH1 and
PMS2. These proteins form two pairs of heterodimers which are obligatory partners.
MSH2 is paired with
MSH6 which recognizes and binds to single base mismatched pairs or insertion-deletion loops in newly synthesized DNA. The second pair of MMR proteins,
MLH1 and
PMS2, joins the complex and recruits an endonuclease to excise the defective daughter strand of DNA. It recruits DNA polymerase to generate a new, correct complementary daughter strain to repair the mismatched DNA
. Mutations of MMR genes result in impaired DNA repair of mononucleotide and dinucleotide repeats throughout the genome and these regions are known as microsatellites. These mutations result in microsatellite instability (MSI). There is altered repeat size with shortened length of allele in the tumour DNA. This difference in length can be detected by polymerase chain reaction (PCR). In the setting of Lynch Syndrome, microsatellite instability is established according to the Bethesda guidelines by testing a simplified panel of five microsatellite loci. These include two mononucleotide repeats: BAT25 and BAT26, and 3 dinucleotide repeats: D2S123, D5S346 and D17S250. If ≥2 of the 5 markers show microsatellite instability, this is classified as MSI high category. If only one marker shows microsatellite instability, this is classified as MSI low category. If none of the markers show microsatellite instability, this is classified as MSI stable
.
1.1. Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome
Lynch syndrome (LS) and Constitutional Mismatch Repair Deficiency (CMMR-D) syndrome arise due to mutations in the MMR genes. They are both inherited tumour predisposition syndromes that manifest with gastrointestinal polyps and develop into early onset colorectal carcinomas. LS is well known to clinicians and pathologists and routine screening and testing for LS-associated colorectal carcinomas are implemented in most diagnostic laboratories. There is research and development into screening and testing of LS-associated breast carcinomas, endometrial carcinomas and other tumours. CMMR-D syndrome is a rare and relatively recently recognized entity in comparison to LS. In order to understand CMMR-D syndrome, we need to discuss the concepts around screening and diagnosis of LS. The differences are discussed and then summarized in
Table 3.
1.1.1. Lynch Syndrome
Lynch syndrome, formerly known as hereditary nonpolyposis colorectal cancer (HNPCC), is a genetic disorder with an autosomal dominant pattern of inheritance. There is an inherited germline mutation of one of the four MMR genes. The remaining wild-type MMR allele undergoes somatic point mutations or loss-of-heterozygosity. This results in frameshift and downstream nonsense mutations that express truncated, non-functional MMR proteins and this impairs the MMR DNA Repair system. Errors in replicating DNA are not repaired and there is an accumulation of spontaneous, single base-pair mismatches with point mutations and insertion-deletion loops. This leads to loss of control of cell growth and cell differentiation, which results in the development of specific types of cancers. LS patients are predisposed to the development of many types of cancers, predominantly colorectal carcinomas and endometrial carcinomas. The Amsterdam and Bethesda criteria are tools used to identify at-risk patients for LS testing
.
1.1.2. Constitutional Mismatch Repair Deficiency Syndrome
CMMR-D syndrome occurs when a patient inherits mutations in both alleles involving the same MMR gene, one from each parent. In half of cases, each parent contributes an allele with a different defect in the same MMR gene and this is known as compound heterozygous mutations. In the other half of cases, each parent contributes an allele with an identical mutation in the same MMR gene and this is known as homozygous mutations. Homozygous mutations typically occur in children of LS-affected families as a result of consanguineous unions
.
Counterintuitively, in spite of the genetic basis of the CMMR-D syndrome, both parents may be unaffected by LS. Family members of the index patient with CMMR-D syndrome do not usually meet the clinical criteria (Amsterdam Criteria) for LS and they are rarely affected by LS-associated cancers
. Lack of awareness around this feature may result in delays in diagnosis. One reason for the lack of parental expression of LS is that 60% of CMMR-D patients show biallelic germline mutations in
PMS2 and the remaining cases show mutations in
MLH1,
MSH2 and MSH6. The penetrance in monoallelic
PMS2 and monoallelic
MSH6 mutated obligate carriers is less than that for other MMR genes
| [14] | Tabori, U, JR Hansford, MI Achatz, CP Kratz, SE Plon, T Frebourg, and L Brugieres. 2017. “Clinical management and tumour surveillance recommendations of inherited mismatch repair deficiency in childhood.” Clinical Cancer Research 23: 32-37. https://doi.org/10.1158/1078-0432.CCR-17-0574 |
[14]
. The reason for this is still unclear, but it may be because these mutations result in only partial impairment of the PMS2 protein
. This is in contrast to
MLH1 and
MSH2 mutations which are identified in the majority of patients with LS.
CMMR-D syndrome is clinically distinct from LS and the patients are at higher risk for developing childhood-onset gastrointestinal cancers and extra-intestinal cancers when compared to other childhood cancer syndromes. The original constellation of features was colorectal cancers, brain tumours, lymphoma and leukaemia with neurofibromatosis type 1-like cutaneous lesions or café au lait-like cutaneous lesions
| [6] | Durno, C, CR Boland, S Cohen, JA Dominitz, FM Giardiello, DA Johnson, T Kaltenbach, et al. 2017. “Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer.” Gastroenterology 152: 1605-1614. https://doi.org/10.1053/j.gastro.2017.02.011 |
[6]
. CMMR-D syndrome patients can develop metachronous gastrointestinal cancers, commonly colorectal or small intestinal carcinomas with associated adenomatous polyps. The number of adenomatous polyps range from 1 to 50 polyps which can make it difficult to distinguish from other polyposis syndromes such as attenuated Familial Adenomatous Polyposis (FAP)
. CMMR-D syndrome patients often are initially presumed to have FAP. When
APC mutations cannot be identified, these patients should be re-evaluated for CMMR-D syndrome
| [6] | Durno, C, CR Boland, S Cohen, JA Dominitz, FM Giardiello, DA Johnson, T Kaltenbach, et al. 2017. “Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer.” Gastroenterology 152: 1605-1614. https://doi.org/10.1053/j.gastro.2017.02.011 |
[6]
. Colorectal carcinomas manifest in adolescence or early adulthood and multiple primary synchronous colorectal carcinomas can also occur. It is important to identify cases of CMMR-D syndrome because it has the highest lifetime risk for gastrointestinal cancers with accelerated progression from adenoma to carcinoma.
POLE and
POLD1 mutations are acquired early and when paired together with MMR deficiency, it results in tumours with massive numbers of substitution mutations and an accelerated rate of progression. CMMR-D syndrome also predisposes to other gastrointestinal and extra-gastrointestinal cancers. 40% of patients will develop a secondary metachronous malignancy
| [13] | Ramchander, NC, NAJ Ryan, EJ Crosbie, and DG Evans. 2017. “Homozygous germ-linemutation of the PMS2 mismatch repair gene: a unique case report of constitutional mismatch repair deficiency (CMMR-D).” BMC Medical Genetics. 18(40). https://doi.org/10.1186/s12881-017-0391-x |
[13]
. Small intestinal carcinoma is rare in childhood; however, CMMR-D patients are at increased risk and there have been reported cases of small intestinal carcinoma in children from as young as 11 years of age
.
1.1.3. The European Consortium’s Care for CMMRD (C4CMMRD)
In CMMR-D syndrome, diagnostic difficulties result from the diverse and wide range of neoplasms and the absence of syndrome-specific clinical features. It is a rare entity and the lack of awareness around it contributes to under-diagnosis and underreporting of cases. This results in missed opportunities to screen the index patient and their families for preventable and treatable cancers. A European consortium, ‘Care for CMMR-D’ (C4CMMR-D), was established and they held a workshop in 2013 to address these issues. Wimmer
et al. characterized the malignancies of 146 patients with CMMR-D syndrome. They developed the criteria for CMMR-D testing in cancer patients, which is outlined in
Table 1.
Table 1. Indications for CMMR-D testing in cancer patients, adapted from Wimmer et al (2014).
Indication for CMMRD testing in a cancer patient ≥3 points |
Malignancies/premalignancies: one is mandatory; if more than one is present in the patient, add the points |
Carcinoma from the LS spectrum* at age <25 years | 3 points |
Multiple bowel adenomas at age <25 years and absence of APC/MUTYH mutation(s) or a single high-grade dysplasia adenoma at age <25 years | 3 points |
WHO grade III or IV glioma at age <25 years | 2 points |
NHL of T-cell lineage or supratentorial embryonal tumours at age <18 years | 2 points |
Any malignancy at age <18 years | 1 point |
Additional features: optional; if more than one of the following is present, add the points |
Clinical sign of NF1 and/or ≥hyperpigmented and/or hypopigmented skin alterations >1 cm in the patient | 2 points |
Diagnosis of LS in a first-degree or second-degree relative | 2 points |
Carcinoma from the LS spectrum* before the age of 60 in first-degree, second-degree and third-degree relative | 1 point |
A sibling with carcinoma from the LS spectrum*, high-grade glioma, supratentorial embryonal tumours or NHL | 2 points |
A sibling with any type of childhood malignancy | 1 point |
Multiple pilomatricomas in the patient | 2 points |
One pilomatricoma in the patient | 1 point |
Agenesis of the corpus callosum or non-therapy-induced cavernoma in the patient | 1 point |
Consanguineous parents | 1 point |
Deficiency/reduced levels of IgG2/4 and/or IgA | 1 point |
*Colorectal, endometrial, small bowel, ureter, renal pelvis, biliary tract, stomach, bladder carcinoma. |
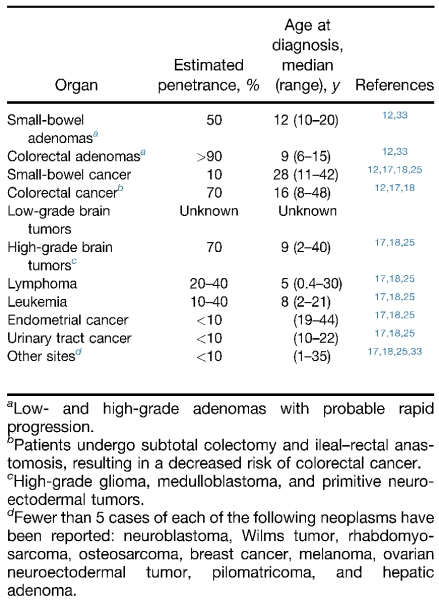
Figure 1. Estimated Penetrance and Age of Onset of Neoplasms in BMMRD (CMMR-D syndrome), adapted from Durno et al (2017).
The tumours associated with CMMR-D syndrome were characterized. 45 patients developed haematolymphoid malignancies, the vast majority were non-Hodgkin lymphomas of T-cell lineage, but also included non-Hodgkin lymphomas of B-cell lineage. Three patients showed two different metachronous haematolymphoid malignancies. The study did not specify the histologic type of lymphomas. 78 patients developed brain tumours, the majority were high grade gliomas including glioblastoma (WHO grade IV) and anaplastic astrocytoma (WHO grade III) tumours. Oligodendrogliomas were not as common, but were also described. 59 patients developed LS-associated malignancies. The majority were colorectal carcinomas. 16 patients developed synchronous colorectal carcinomas. Other LS-associated tumours included small bowel carcinomas, endometrial carcinoma, urethral carcinoma and ovarian carcinoma. Of the other tumours, one of each kind was represented including neuroblastoma, nephroblastoma, ovarian neuroectodermal tumour, infantile myofibromatosis, rhabdomyosarcoma, basal cell carcinoma, mucoepidermoid carcinoma of the parotid gland and osteosarcoma. None of these tumours are unique to CMMR-D syndrome and some may not be related to the underlying mutation. However, suspicion of an underlying tumour syndrome may be raised if multiple tumours occur especially at a young age at presentation (See
Figure 1).
Neurofibromatosis type 1 (NF1) -like features have been observed in most, but not all patients with CMMR-D syndrome. In contrast to CMMR-D syndrome, NF1 is an autosomal dominant disorder which results from mutations in the
Neurofibromin 1 gene. Café-au-lait spots are the main cutaneous manifestation of NF1. Patients with CMMR-D syndrome-associated NF1 lesions have no family history of NF1. It has been suggested that the NF1-like manifestations result as a consequence of the failure in the DNA repair mechanism
.
1.2. Laboratory Diagnosis of CMMR-D Syndrome
1.2.1. MMR Immunohistochemistry
Immunohistochemical (IHC) stains may be applied to formalin fixed paraffin embedded (FFPE) tissue sections to identify loss of expression of each of the four MMR proteins. This method is already in use in most diagnostic laboratories because there are established screening protocols for LS in colorectal carcinomas
. Generally, MMR immunohistochemistry is widely available, more cost effective and has better turnaround times when compared to PCR testing or other molecular techniques.
1.2.2. Use of MMR Immunohistochemistry in Lynch Syndrome
In LS, the Bethesda criteria and the Amsterdam criteria are used to identify patients for screening by immunohistochemistry. Cases that have intact nuclear staining of all four MMR proteins are generally excluded from the pool of suspected LS cases. LS patients with missense mutations can show intact staining of all four MMR immunostains, and this is a recognized cause for a false negative result. Diagnosis in these cases depends on strong clinical suspicion and referral for further testing by molecular techniques
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
. In the evaluation of MMR immunohistochemistry for LS screening, both the tumour cells and the background cells are evaluated. In tumour cells, any nuclear staining is considered positive
| [2] | Bartley, AN, SR Hamilton, R Alsabeh, EP Ambinder, M Berman, E Collins, PL Fitzgibbons, et al. 2014. “Template for Reporting Results of Biomarker Testing of Specimens From Patients with Carcinoma of the Colon and Rectum.” December. Accessed June 2019. |
[2]
; however, careful evaluation is required because the tumour can show heterogenous staining with patchy loss of nuclear staining. The background, non-tumour colonic epithelial cells, stromal cells, tumour infiltrating lymphocytes and inflammatory cells are expected to have intact nuclear positive staining and they are used as inbuilt positive controls
| [2] | Bartley, AN, SR Hamilton, R Alsabeh, EP Ambinder, M Berman, E Collins, PL Fitzgibbons, et al. 2014. “Template for Reporting Results of Biomarker Testing of Specimens From Patients with Carcinoma of the Colon and Rectum.” December. Accessed June 2019. |
[2]
. In cases that show loss of immunostaining, the pattern of staining allows for targeted molecular genetic testing. Instead of testing all four MMR genes, we can start with the single MMR gene, or the dominant heterodimer that showed loss of immunostaining. Mutations in
PMS2 or
MSH6 result in isolated loss of these proteins, whereas mutations in
MLH1 or
MSH2 leads to concurrent loss of
MLH1/
PMS2 or
MSH2/
MSH6, respectively, since
MLH1 and
MSH2 are the obligatory partners
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
. The pattern of loss of staining in the tumour cells between the four MMR proteins is used to stratify patients into MSI stable tumours and tumours that require further investigations. Diagnostic algorithms are applied to exclude loss of immunostaining caused by other mechanisms such as MLH1 loss due to promoter hypermethylation
.
1.2.3. Use of MMR Immunohistochemistry in CMMR-D Syndrome
In the setting of CMMR-D syndrome, immunohistochemistry is proposed as a diagnostic tool due to the unique staining patterns which was found to be reliable in the evaluation of solid tumours. In contrast to LS, CMMR-D patients show loss of expression of the affected MMR gene in both tumour cells and in the background non-tumour tissue. In general, if this occurs with non-MMR immunohistochemical stains, it prompts checking of the external positive control. If it has failed, the test is considered inadequate and it usually requires review of preanalytical factors with repeat application of staining. In the setting of CMMR-D syndrome, this pattern of staining should not be interpreted as failure of the staining, provided that the external positive control is adequate
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
. In patients with suspected CMMR-D syndrome, if there is no tumour material available for testing, a normal skin sample may be submitted for testing to demonstrate the loss of staining in normal tissue
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
. Genetic testing on blood is also possible.
In the C4CMMRD guidelines and review of the literature of the use of MMR immunohistochemistry in CMMR-D syndrome, there is no explanation or characterization of what is considered loss of staining in tumour cells. It is unclear if the same scoring conventions of MMR immunohistochemistry for Lynch syndrome can be applied in CMMR-D syndrome. It is unclear if reduced staining may be considered a negative staining pattern.
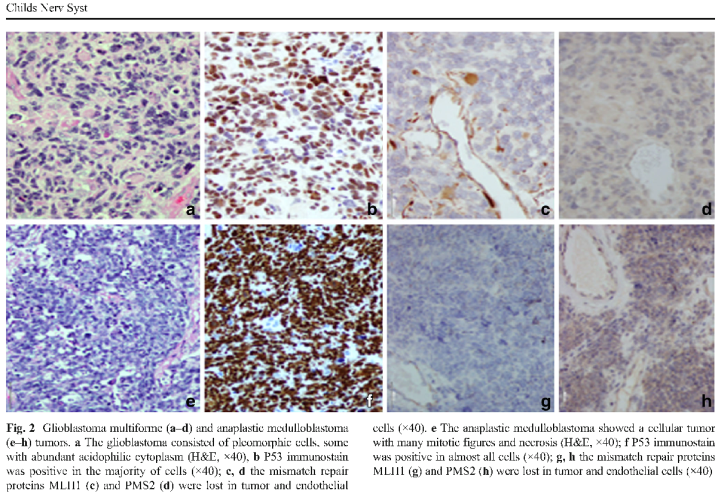
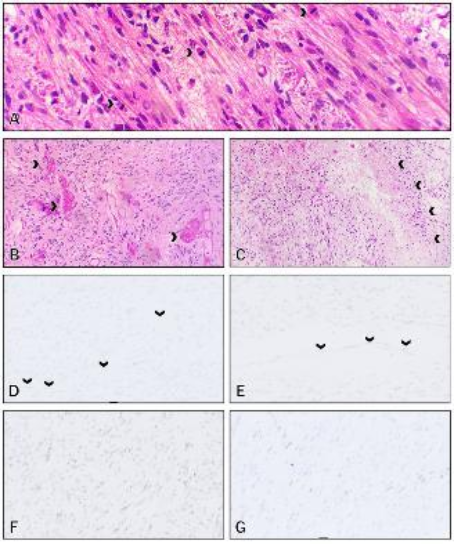
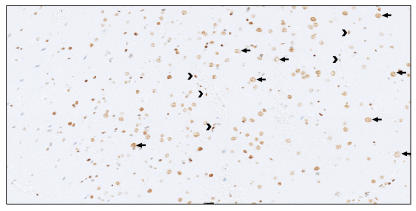
Figure 2. A figure from Amayiri et al. (2016) describing loss of staining in endothelial cells; however, positive endothelial staining is seen in (c) and (h).
Loss of MMR immunostaining in background non-tumour tissue is characteristic in CMMR-D syndrome; however, there is a problem with variable reporting of what is considered normal tissue and/or lack of documentation of which normal tissue were or should be evaluated. The tissue types mentioned include lymphocytes within blood vessels, tumour infiltrating lymphocytes, endothelial cells and “normal tissue”. One article even describes loss of staining in endothelial cells, then shows an image of positive staining of endothelial cells (See
Figure 2). This is a critical aspect for the interpretation of MMR immunohistochemistry staining, and it should be described with more consistency, especially considering the implications of such a result. In high grade gliomas, the prognosis is worse in patients with CMMR-D syndrome
| [4] | Carrato C, C Sanz, AM Muñoz-Mármol, I Blanco, M Pineda, J Del Valle, E Dámaso, M Esteller, E Musulen. 2021. The Challenge of Diagnosing Constitutional Mismatch Repair Deficiency Syndrome in Brain Malignancies from Young Individuals. The Journal of Molecular Sciences. 22(9): 4629. https://doi.org/10.3390/ijms22094629 |
[4]
. MMR deficient tumours are responsive to immune checkpoint inhibitors, specifically gastrointestinal and brain tumours. MMR deficient tumours are resistant to chemotherapies, such as temozolomide, which is commonly used to treat brain tumours
| [10] | Kang, E, JK Suh and SD Kim. 2025. “Constitutional Mismatch Repair Deficiency, the Most Aggressive Cancer Predisposition Syndrome: Clinical Presentation, Surveillance, and Management”. Journal of Korean Neurosurgical Society 68(3): 294–304. https://doi.org/10.3340/jkns.2025.0024 |
[10]
. If CMMR-D syndrome is diagnosed, the ERN GENTURIS guidelines developed surveillance recommendations for the early detection of tumours in affected individuals
| [5] | Colas, C, L Guerrini-Rousseau, M Suerink, R Gallon, CP Kratz, É Ayuso, ERN GENTURIS CMMRD Guideline Group, L Brugières, K Wimmer. 2024. “ERN GENTURIS guidelines on constitutional mismatch repair deficiency diagnosis, genetic counselling, surveillance, quality of life, and clinical management”. European Journal of Human Genetics. 32(12): 526–1541. https://doi.org/10.1038/s41431-024-01708-6 |
[5]
. Some patients with CMMRD have multiple metachronous cancers. They may exceed the maximum tolerated lifetime dose limits for radiation and certain chemotherapy agents. Surveillance enables these patients to be managed with surgery and follow-up alone
| [7] | Durno C, AB Ercan, V Bianchi, M Edwards, M Aronson, M Galati, EG Atenafu, On Behalf of the International Replication Repair Deficiency Consortium. 2021. Survival Benefit for Individuals With Constitutional Mismatch Repair Deficiency Undergoing Surveillance. Journal of Clinical Oncology. Volume 39, Number 25. https://doi.org/10.1200/JCO.20.02636 |
[7]
.
1.2.4. Objective and Reproducible Reporting of Immunohistochemical Stains in General
In studies that involve the evaluation of staining of immunohistochemical stains, including MMR immunohistochemistry, it is important to evaluate and report the stains in an objective manner. This standardization allows for comparison of the findings between different individual samples, different tumour types and different study cohorts. Fedchencko
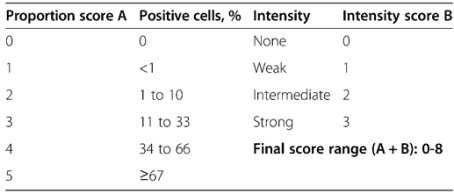
et al. outline semiquantitative scoring systems which are used to standardize the evaluation of immunohistochemical stains and reduce inter-observer variability. Parameters, such as intensity of staining and proportion of stained tumour cells, are evaluated and converted into quantitative data, which is then used for statistical analyses. The “gold standard” in the scoring of immunohistochemistry is the evaluation of oestrogen receptor, progesterone receptor and Her2/neu in breast tumour specimens. The Allred scoring system is widely accepted and used by leading associations and organizations. The percentage of positive cells is scored and the intensity of reaction in most examined fields is scored, and the cut off values are outlined in
Figure 3. The scores are then added together for a final score that may range from 0 to 8. In this system, scores of 0 and 2 are considered negative, and scores of 3 to 8 are considered positive
| [8] | Fedchenko, N, and J Reifenrath. 2014. “Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue - a review.” Diagnostic Pathology (9): 221. https://doi.org/10.1186/s13000-014-0221-9 |
[8]
.
Figure 3. Allred scoring system outlined in Fedchenko et al. (2014).
1.2.5. Microsatellite Instability Testing in CMMR-D Syndrome
MSI analysis with current protocols for LS is extremely sensitive and specific for LS-associated tumours, but it may be negative in CMMR-D cancers, especially in extraintestinal cancers
| [14] | Tabori, U, JR Hansford, MI Achatz, CP Kratz, SE Plon, T Frebourg, and L Brugieres. 2017. “Clinical management and tumour surveillance recommendations of inherited mismatch repair deficiency in childhood.” Clinical Cancer Research 23: 32-37. https://doi.org/10.1158/1078-0432.CCR-17-0574 |
[14]
. It is not recommended as a screening tool because MSI status may be unreliable in brain tumours and haematological malignancies for reasons that are not yet known
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
. This may be because they diffusely infiltrate into adjacent tissue and testing normal tissue is difficult. Different MSI genotypes can be observed in different tumour tissue in patients affected by multiple cancers. MSI status may be tumour specific and not related to the mutation
. The reason for this is unknown, but there are subtler shifts of microsatellite alleles observed in brain tumours. A different panel of microsatellite markers could be more sensitive for brain tumours and other tumours in the setting of CMMR-D syndrome.
According to Wimmer
et al. a recent, simpler assay analysing “stutter” peaks was presented for the detection of MSI in CMMR-D syndrome patients. Stutter peaks are associated with microsatellite PCR products. A new, publically available software application is used to quantify the peak height of the “stutter” peaks in selected dinucleotide microsatellites. In CMMR-D syndrome patients, this peak significantly increases compared to normal controls, but is limited to
PMS2,
MLH1 and
MSH2 mutations. The peak height in patients with biallelic
MSH6 mutations is not altered
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
.
1.2.6. Molecular Genetic Testing
Genetic testing is recommended to help confirm the diagnosis of CMMR-D syndrome. It is used to detect biallelic mutations in one of four MMR genes. These genes have many variants of unknown significance. The
PMS2 gene also has multiple pseudogenes which makes it difficult to sequence
| [14] | Tabori, U, JR Hansford, MI Achatz, CP Kratz, SE Plon, T Frebourg, and L Brugieres. 2017. “Clinical management and tumour surveillance recommendations of inherited mismatch repair deficiency in childhood.” Clinical Cancer Research 23: 32-37. https://doi.org/10.1158/1078-0432.CCR-17-0574 |
[14]
.
Tabori
et al. suggest that MSI should not be used as a diagnostic tool for CMMR-D syndrome because of the risk of false negative results and that immunohistochemical stains should be used as a screening tool on any index patients and family members
| [14] | Tabori, U, JR Hansford, MI Achatz, CP Kratz, SE Plon, T Frebourg, and L Brugieres. 2017. “Clinical management and tumour surveillance recommendations of inherited mismatch repair deficiency in childhood.” Clinical Cancer Research 23: 32-37. https://doi.org/10.1158/1078-0432.CCR-17-0574 |
[14]
.
In the C4CMMRD guidelines, it is acknowledged that both immunohistochemistry and MSI testing have potential pitfalls. They recommend a combination of both methods if needed. Final confirmation comes from the identification of the causative biallelic mutation
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
.
1.3. Relevance to the Lynch Syndrome Families of Cape Town
There is a well-known population of LS affected families in the Western Cape and Northern Cape Provinces of South Africa. They are regularly followed up by clinicians and clinical geneticists as part of the University of Cape Town (UCT) Human Genetics registry. LS populations across the world can show varied predominance of the affected MMR gene and specific mutations. In the South African population, the MLH1 c1528C>T mutation is the most common mutation. It is unique to the mixed ancestry population and is recognized as a founder mutation. The mutation aging analysis traced this mutation to be an estimated 225 years old (1789 – 1814); however, the “founder” of this founder effect in this population has not yet been identified. The founder effect may explain the higher frequency of LS in the Cape mixed ancestry populations of the Western and Northern Cape provinces compared to the rest of South Africa
.
Consanguinity and founder effect are the two mechanisms that contribute to the development of CMMR-D syndrome
| [13] | Ramchander, NC, NAJ Ryan, EJ Crosbie, and DG Evans. 2017. “Homozygous germ-linemutation of the PMS2 mismatch repair gene: a unique case report of constitutional mismatch repair deficiency (CMMR-D).” BMC Medical Genetics. 18(40). https://doi.org/10.1186/s12881-017-0391-x |
[13]
. Mutations in both
MLH1 and
MSH2 contribute to the entire cohort of LS cases in the University of Cape Town (UCT) Human Genetics registry. This is a concern because CMMR-D syndrome arising from mutations in
MLH1/
MSH2 result in a more severe phenotype. The percentage of biallelic mutation carriers with more than one malignancy is lowest in
MLH1/
MSH2 carriers (22%) and highest in
PMS2 biallelics (42%)
| [15] | Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365. https://doi.org/10.1136/jmedgenet-2014-102284 |
[15]
. This may indicate that patients with PMS2 biallelic patients are more likely to survive to develop a second metachronous tumour, whereas those with
MLH1/
PMS2 are less likely to survive to develop a second metachronous tumour.
The incidence of CMMR-D syndrome may be different in our population because the majority of cases of CMMR-D syndrome are attributed to mutations in
PMS2, but there is a predominance of MLH1/MSH2 mutations in our population.
Table 2 shows the average age of onset of different tumours in biallelic
MLH1 carrier cases.
Table 2. Average age of malignancy presentation / detection amoung reported biallelic MLH1 mutation carrier cases, adapted from Bruwer et al. (2014).
Site of malignancy | Average age of onset for specific malignancy |
Haematological | 2.9 years |
Brain | 4.5 years |
Colorectal | 17.8 years |
Mean age of onset of any malignancy | 8.4 years |
Range | 1 year to 35 years |
In contrast to the founder effect seen in the South African population, a study in Jordan looked at a region with high rates of consanguinity. They found that there is a higher prevalence of CMMR-D syndrome compared to a North American cohort. They found that up to 51% of childhood gliomas in Jordan may harbour germline mutations in MMR genes
| [1] | Amayiri, N, U Tabori, B Campbell, D Bakry, M Aronson, C Durno, P Rakopoulos, et al. 2016. “High frequency of mismatch repair deficiency among pediatric high grade gliomas in Jordan.” International Journal of Cancer 138: 380-385. https://doi.org/10.1002/ijc.29724 |
[1]
.
The first published report of predictive genetic testing for CMMR-D syndrome came from our institution and the University of Cape Town (UCT) Human Genetics registry. Both the mother and father showed heterozygous mutation in
MLH1 c1528C>T, the most common mutation in our population. The parents were of mixed ancestry and they were not related to each other. They had two daughters and the parents were offered predictive testing and genetic counselling due to the concern from CMMR-D syndrome. The older daughter had café au lait spots and was tested and confirmed to have biallelic mutations in
MLH1 c1528C>T, CMMR-D syndrome. She later died from complications of a high-grade diffuse astrocytoma of the brain stem, cerebellum and thalamus despite normal screening tests. The brain MRI and blood work were normal six months prior to the development of symptoms. The younger daughter was also tested and showed heterozygous mutation in
MLH1 c1528C>T
| [3] | Bruwer, Z, U Algar, A Vorster, K Fieggen, A Davidson, P Goldberg, H Wainwright, and R Ramesar. 2014. “Predictive genetic testing in children: Constitutional mismatch repair deficiency cancer predisposing syndrome.” Journal of Genetic Counselling 23: 147-155. https://doi.org/10.1007/s10897-013-9659-2 |
[3]
.
Wimmer et al. characterized the expression of tumours in patients with an established diagnosis of CMMR-D syndrome. They suggested that there is an under-recognition and under-diagnosis of CMMR-D syndrome because there is a varied presentation, overlapping features with other tumour syndromes such as NF1 and FAP and lack of specific features of this syndrome. The diagnosis depends on increased awareness and a higher index of suspicion to avoid a missed or a delayed diagnosis. This study aimed to address this issue. When paediatricians identify a patient with clinical suspicion for CMMR-D syndrome, by applying MMR immunohistochemistry to tumours that fit part of the CMMR-D syndrome spectrum, can we identify cases that are MMR deficient? Is there a role of the pathologist to flag these cases for the attention of the clinicians and trigger genetic testing and genetic counselling of affected families? This may be different in the South African population because we have established LS families with a unique MLH1 mutation and potentially a more aggressive phenotype.
Table 3. Comparison of Lynch Syndrome and CMMR-D Syndrome.
Lynch Syndrome | | CMMR-D Syndrome |
GENETICS |
Autosomal dominant | Inheritance pattern | Autosomal recessive |
Founder effect | Mechanism of inheritance | Founder effect and/or consanguineous unions |
Germline mutation in an MMR gene with second acquired somatic mutation | Type of mutation | Biallelic germline mutation in the same MMR gene |
MLH1 and MSH2 genes Unique MLH1 c1528C>T mutation in mixed ancestry population in South Africa | Predominantly mutated MMR genes | PMS2 and MSH6 genes |
CLINICAL FINDINGS |
Expected autosomal dominant pedigrees | Family history of Lynch Syndrome | May not be present. PMS2 mutations have partial penetrance, due to possible partial inactivation |
± 50 years | Average age of onset of MMR-deficient colorectal carcinomas | 17.8 years |
Not classically described | Cutaneous manifestations | Café-au-lait-like macules and multiple pilomatrixomas |
Breast carcinoma and endometrial carcinoma | Commonly associated extraintestinal tumours | High grade gliomas, Non-Hodgkin T-cell lymphoma, leukaemia |
Amsterdam and Bethesda criteria | Tools for evaluation of at-risk patients | C4CMMRD criteria |
MMR IMMUNOHISTOCHEMISTRY |
Established protocols for routine use in colorectal carcinomas | Use as a screening tool | Some conflicting recommendations. Most recommend MMR IHC as the first investigation, to direct targeted molecular testing |
Mutated MMR gene ±heterodimer: Loss of nuclear staining in tumour cells & Non-mutated genes Intact nuclear staining in tumour cells | Expected staining in tumour cells (patients with MMR deficiency) | Mutated MMR gene ±heterodimer: Loss of nuclear staining in tumour cells & Non-mutated genes: Intact nuclear staining in tumour cells |
In both mutated and non-mutated genes: Intact nuclear staining in stromal cells, inflammatory cells and non-tumour epithelial cells (established for colorectal carcinomas) Used as an inbuilt positive control | Expected staining in background tissue (patients with MMR deficiency) | Mutated MMR genes ±heterodimer: Loss of nuclear staining in background tissue At risk of being interpreted as failed staining & Non-mutated genes: Intact nuclear staining in background tissue |
Colorectal carcinomas: MLH1 loss can also be seen in tumours with promotor hypermethylation (false positive) Lynch syndrome with missense mutations can show falsely intact staining (false negative) | Recognized causes for false results | Missense mutations can show falsely intact nuclear staining (false negative) |
MSI LOCUS TESTING |
MMR immunohistochemistry determines if MSI testing is recommended Extremely sensitive and specific for Lynch syndrome associated tumours | MSI locus testing | Not recommended as a screening tool. May be unreliable in brain tumours and haematological malignancies. Different MSI genotypes can be observed in different tumour tissue in patients affected by multiple cancers |
MOLECULAR GENETIC TESTING |
Gold standard. Immunohistochemistry can identify which gene to test. Recommended if there are conflicting results from immunohistochemistry and MSI testing | Molecular genetic testing | Immunohistochemistry can identify which gene to test. Recommended to use IHC in conjunction with molecular genetic testing |
2. Materials and Methods
2.1. Design and Setting
This study was a retrospective study using archival formalin fixed, paraffin embedded (FFPE) tissue samples of paediatric patients attending Red Cross Children’s Hospital. The C4CMMR-D criteria, outlined by Wimmer et al., was used to identify at-risk patients. MMR immunohistochemical stains were applied to tissue sections of each case: MLH1, PMS2, MSH2 and MSH6. Decreased or absent staining of one or more of these stains indicated abnormalities in mismatch repair protein expression and the possible diagnosis of CMMR-D syndrome. These cases will be referred to the Department of Human Genetics for genetic counselling and proposed confirmatory testing.
2.2. Sampling
A clinical search was performed to identify paediatric oncology patients with a family history of LS, patients with cutaneous lesions suggestive of CMMR-D syndrome and patients diagnosed with tumours in the spectrum of CMMR-D syndrome. A computerised search of the entire Red Cross Hospital histopathology database was performed to retrieve relevant cases from 1988 to 2014.
A modified version of Wimmer
et al. 's “
Indication for CMMRD testing in a cancer patient” criteria (See
Table 1) to determine the types of CMMR-D associated tumours to search. The search included patients with one or more of the following conditions or neoplasms:
Endometrial, colorectal, small bowel, ureter, renal pelvis, biliary tract, stomach and bladder carcinoma diagnosed <25 years of age (LS spectrum carcinomas)
Multiple bowel adenomas / single high-grade adenoma, < 25 years of age
WHO grade III or IV gliomas, <25 years of age
Non-Hodgkin T-cell lymphoma or supratentorial primitive neuroectodermal tumour <18 years of age
Any malignancy at age <18 years with café au lait spots (≥2 hyperpigmented and/or hypopigmented skin macules
Multiple pilomatrixomas
Agenesis of corpus callosum or non-treatment related cavernoma
Deficiency/reduced levels of IgG2/4 and/or IgA
Surgical biopsies, frozen section samples and post-mortem samples were included. Cytologic specimens, samples with insufficient viable tumour material and adult samples were excluded. The samples included in this study are from patients with suspected CMMR-D syndrome and have not been genetically confirmed.
2.3. Study Procedures
Each sample was assigned a unique study number to protect patients’ identifying details. The electronic documents used to capture this information were password protected and access was limited to the study staff only. The patients’ age, diagnosis, date and site of sample were documented. A selected folder review of cases with features compatible with CMMR-D syndrome was performed.
The Haematoxylin and Eosin (H&E) slides of each sample were evaluated, and the best slide was selected to minimize factors that would interfere with the staining process, such as excessive tumour necrosis and poor tissue preservation. The corresponding tissue blocks were retrieved from archives. Additional sections were made, and each sample was stained with the four immunohistochemical stains: PMS2, MLH1, MSH2 and MSH6.
2.4. Methodology
Immunohistochemistry was performed using the commercially-available, automated detection system – Roche Optiview DAB detection system and automated supplier antibodies. The preparation of all buffers and reagents used in the staining protocol are listed in Appendix 1.
Three-micron FFPE tissue sections were cut on a Leica Rotary Microtome RM2125 RTS. The sections were floated on a water bath and picked up on coated Histobond slides. The sections were then heat fixed on a hotplate at 60°C for 10-15 minutes.
Thereafter, the various MMR antibody protocols were selected on the auto-stainer, and a barcoded slide label with the study number and date was generated.
The system generated label was fixed to the slide and then loaded onto the auto-stainer. The required bulk reagents were re-constituted with water (Appendix 1), loaded, and registered on the auto-stainer. The detection system was also loaded and registered on the auto stainer. The run was then started, and a brief dewaxing took place using EZPREP. Antigen retrieval, using CC1 was then performed for 64 minutes. After antigen retrieval, each of the primary antibodies was applied automatically, and incubation took place for 36 minutes. The Optiview DAB detection kit reagents were then applied to the slides to visualise the antigen/antibody complex. The slides were then automatically counterstained with haematoxylin and blued using bluing reagent. When the staining protocol had run to completion, the slides were removed from the auto-stainer and washed briefly in diluted washing up detergent and water in order to remove the LCS.
The sections were dehydrated in alcohol, cleared in xylene, and then the slides were cover slipped using glass coverslips and Entellan.
2.5. Outcome Measures
Evaluation of Immunohistochemical Stains
The MMR staining was evaluated by Dr S Tu on an Olympus BX43 microscope. A modified Allred method of scoring immunohistochemistry was used to quantify the proportion of viable tumour cells that showed positive nuclear staining and the intensity of the staining. Any nuclear staining of tumour cells is considered positive staining for MMR immunohistochemistry in the setting of Lynch Syndrome
| [2] | Bartley, AN, SR Hamilton, R Alsabeh, EP Ambinder, M Berman, E Collins, PL Fitzgibbons, et al. 2014. “Template for Reporting Results of Biomarker Testing of Specimens From Patients with Carcinoma of the Colon and Rectum.” December. Accessed June 2019. |
[2]
.
In this study, the Allred method (See
Figure 3) is used to record the intensity and proportion of each sample in an objective manner. The system is modified to exclude the final score because we do not know yet which scores are significant. Comments on staining of the background tissue were also included. These results were documented on a password protected electronic spread sheet.
3. Results
A total of 21 samples were selected from 18 patients. Two patients had two samples that were taken at the same instance with concordant diagnoses. One patient had two samples taken at an interval of 6 months with concordant diagnoses.
A total of four post-mortem samples were evaluated, with an average post-mortem interval of 1.75 days and a range of 1 to 4 days. One sample was from an intraoperative frozen section and the remaining 16 samples were routine surgical biopsies.
20 samples were submitted to the laboratory at Red Cross Hospital. One sample was submitted to the laboratory at Frere Hospital, a referral hospital located in East London, Eastern Cape Province and was subsequently sent to Red Cross Children’s Hospital as a referral case.
The ages of the children were converted into months, to allow for fair comparison from infants to older children. Red Cross Children’s Hospital has a cut-off age for admissions of 14 years. The referral case from East London did not include the age or date of birth of the patient. This sample was excluded in the calculation of the average age of patients. The average age of the patients was 82 months (6 years 9 months).
Of the 21 samples taken from 18 patients, the diagnoses were outlined in
Table 4.
Table 4. Histologic diagnosis and anatomic site of the tissue samples.
Organ system | Histologic diagnosis | Site of tumour | Number of cases |
CNS malignancies (16) | Glioblastoma (WHO Grade IV) | Brainstem | 3 (2 cases had 2 samples each) |
| | Frontal lobe | 2 |
| | Thalamus | 3 |
| | Parietal lobe | 1 |
| | Site not specific | 3 |
| Anaplastic astrocytoma (WHO Grade III) | | 2 |
| Low grade oligodendroglioma | | 1 |
| Choroid plexus carcinoma | | 1 |
Haematolymphoid malignancies (1) | Burkitt lymphoma | Left cervical region | 1 (1 case with 2 samples taken at a 1-year interval) |
Other (1) | Anaplastic embryonal rhabdomyosarcoma | Testicular | 1 |
3.1. Evaluation of MMR Immunohistochemical Stains
Each of the four MMR stains had a confirmed positive external control, which excludes failed immunohistochemistry as a cause for negative staining tumour cells. A normal lymph node was used as the control tissue.
Three samples showed suboptimal staining. Samples 2,3 and 4 showed absent to weak nuclear staining in all four MMR immunostains in the tumour cells.
12 samples showed strong, diffuse, intact nuclear staining in all four antibodies in the tumour cells.
Two samples showed staining patterns that are not recognized in CMMR-D syndrome: Sample 19 showed absent or reduced nuclear staining in PMS2 and MSH2. Sample 21 showed absent or reduced nuclear staining in MSH2, PMS2 and MSH6.
Four samples showed staining patterns compatible with CMMR-D syndrome. Sample 16 showed isolated reduced nuclear staining of PMS2. Sample 1 showed reduced staining of MLH1 and PMS2. Sample 8 and 18 were taken from the same patient at a 6-month interval. Both samples showed loss of MSH2 and reduced staining of MSH6.
The scoring of the immunohistochemical staining was summarized in
Table 5. Areas of necrosis, cautery and poorly preserved tumour cells were avoided as these areas are known to show unreliable staining.
Table 5. Modified Allred Scoring for MMR Immunohistochemistry in each Tissue Sample.
| MSH2 | MLH1 | PMS2 | MSH6 |
Case | P | I | P | I | P | I | P | I |
1 | 5 | 2 | 2 | 1 | 1 | 1 | 4 | 2 |
2 | 1 | 1 | 2 | 1 | 1 | 1 | 0 | 0 |
3 | 2 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
4 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
5 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
6 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
7 | 4 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
8 | 0 | 0 | 5 | 3 | 5 | 2 | 2 | 1 |
9 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
10 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
11 | 5 | 3 | 5 | 3 | 4 | 3 | 5 | 3 |
12 | 5 | 3 | 5 | 3 | 4 | 3 | 4 | 3 |
13 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
14 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
15 | 4 | 3 | 5 | 3 | 5 | 3 | 4 | 2 |
16 | 5 | 3 | 5 | 2 | 1 | 1 | 5 | 3 |
17 | 5 | 3 | 5 | 3 | 5 | 3 | 5 | 3 |
18 | 0 | 0 | 5 | 3 | 5 | 3 | 4 | 1 |
19 | 1 | 1 | 5 | 3 | 2 | 2 | 4 | 3 |
20 | 5 | 3 | 5 | 2 | 5 | 2 | 5 | 3 |
21 | 2 | 2 | 5 | 3 | 2 | 3 | 3 | 3 |
Proportion (P) and intensity (I). White cells indicate cases of interest with MMR loss in tumour cells. Grey cells indicate intact MMR staining. Dark grey cells indicate suboptimal staining. |
Comments on the background tissue staining characteristics were included (see
Table 6). The majority of samples had representation of vessels and endothelial cells. In samples of high-grade glioma, the neurons, vessels and choroid plexus showed consistent and strong positive nuclear staining. In lymphomas, endothelial cells showed consistent and strong positive nuclear staining. Tumour infiltrating lymphocytes were difficult to evaluate. In the control tissue of normal lymph node, the germinal centre lymphocytes showed nuclear positivity. The lymphocytes in the interfollicular zone show variable, but predominantly negative nuclear staining. Intravascular, circulating lymphocytes were difficult to identify.
Table 6. Characterization of MMR staining in the background, non-tumour tissue.
| MSH2 | MLH1 | PMS2 | MSH6 |
| Comment | Comment | Comment | Comment |
1 | Negative | Negative | Negative | Positive |
2 | Negative | Negative | Negative | Negative |
3 | Negative | Negative | Negative | Negative |
4 | Positive | Positive | Positive | Positive |
5 | Positive | Positive | Positive | Positive |
6 | Positive | Small fragment | Small fragment | Small fragment |
7 | Positive | Positive | Positive | Positive |
8 | Negative | Positive | Positive | Negative |
9 | Small fragment | Small fragment | Small fragment | Small fragment |
10 | Positive | Positive | Positive | Positive |
11 | Positive | Positive | Positive | Positive |
12 | Small fragment | Positive | Positive | Positive |
13 | Positive | Positive | Positive | Positive |
14 | Positive | Positive | Positive | Positive |
15 | Small fragment | Small fragment | Small fragment | Small fragment |
16 | Positive | Positive | Positive | Positive |
17 | Positive | Positive | Positive | Positive |
18 | Negative | Positive | Positive | Negative |
19 | Negative | Negative | Negative | Positive |
20 | Positive | Positive | Positive | Positive |
21 | Positive | Positive | Positive | Positive |
White cells indicate cases of interest identified in Table 4 and comments on staining pattern of background non-tumour tissue; grey cells indicate remaining cases with intact MMR staining; dark grey cells indicate suboptimal staining in tumour cells |
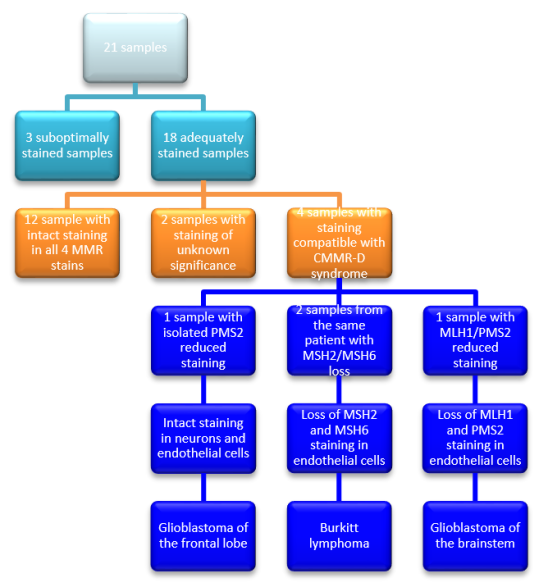
Figure 4 shows the process of elimination of samples that are unlikely to confirm CMMR-D syndrome. The pattern of loss of MMR staining is described in the remaining four samples with comments on the background tissue and the histologic tumour type.
Figure 4. Outcomes of MMR Immunohistochemistry staining with positive external controls.
3.2. Selected Folder Review
A selected folder review was performed on cases with staining patterns compatible with CMMR-D syndrome.
Sample 1:
The patient was a 7-year-old girl who was brought to Red Cross Children’s Hospital by her mother and aunt. She presented with a one week history of difficulty in speech, dysphagia and weakness in the left arm and leg. She was assessed with a left-sided hemiparesis, a right-sided upper motor neuron 7th nerve palsy, a left-sided 6th nerve palsy and a pseudobulbar 10th nerve palsy. There were no skin rashes identified and café au lait-like lesions were not described. Screening tests for HIV and tuberculosis were negative. A brain MRI was performed and showed a poorly enhancing lesion in the brainstem. The largest component was present in the medulla with extension into the pons. The rhomboid fossa showed upward displacement. It was assessed as a diffuse pontine glioma without features of hydrocephalus. In view of the diffuse nature and extent of the lesion, the tumour was not resectable. Her parents were counselled regarding the prognosis and palliative care was provided. She died in hospital, 8 days after admission. Her parents were counselled and consented for a post-mortem examination to be performed. Histology confirmed a high grade pontine glioma. There was no documentation of a genogram or siblings in the hospital notes.
Sample 8 and 18:
The patient was a 2-year-old boy who was referred from a private paediatrician with one and a half week history of a left cervical mass. It was noted that he had “cutaneous neurofibromatosis” at birth without a family history of neurofibromatosis. There was no documentation of confirmatory genetic testing. At Red Cross Oncology, he was assessed with Burkitt Lymphoma on 27/10/1988 (sample 18) and commenced on cyclophosphamide, vincristine, methotrexate, prednisone and allopurinol. He had local and bone marrow recurrence in February 1989 (sample 8). He started on chemotherapy (French protocol) on 06/03/1989. It was documented that there was an uncomplicated induction course. He developed a pseudomonas septicaemia with thrombocytopaenia and neutropenia and a slow resolution on antibiotics. The cervical mass showed initial reduction in size, but started progressively enlarging again. In view of this, it was decided to stop further chemotherapy with continuation of palliative care only. He was discharged on 05/04/1989. There was no documentation of a genogram or siblings in the hospital notes. The oncology folder was reviewed. The medical folder was not found and was documented as destroyed.
Sample 16:
The oncology folder only contained a note that she presented on 16/11/2004 at 3 years of age with neck stiffness and that a debulking craniotomy was performed 2 days after admission. The medical hospital folder was documented as destroyed. In electronic medical records, the date of death was documented as 16/04/2005, 5 months after the date of biopsy confirming a glioblastoma (WHO Grade IV).
4. Discussion
4.1. Suboptimal Staining
Three samples, sample 2, 3 and 4, showed weak staining in all four MMR stains despite adequate positive external controls for all four MMR immunostains (See
Figure 5). All three samples were post-mortem samples and the post-mortem interval may have resulted in autolysis, reduced antigen retrieval and reduced staining across all four antibodies. Sample 2 and 3 were from the same patient with a post-mortem interval of 1 day. Sample 4 had a post-mortem interval of 4 days.
Figure 5. MSH2 control - A normal lymph node used as positive external control tissue.
A: Low power view shows a normal lymph node with preserved follicular architecture.
B: Medium power view shows positive staining of central germinal centre lymphocytes and variable staining of peripheral interfollicular lymphocytes
Sample 1 is the fourth and final post-mortem sample but was evaluated as an adequate sample, to be discussed below under staining patterns compatible with CMMR-D syndrome. Sample 1 had a post-mortem interval of 1 day. Even though it was evaluated as adequate, it is important to note that the MMR proteins that showed intact or retained nuclear staining were patchier with reduced intensity of staining.
Overall, MMR testing of post-mortem specimens by immunohistochemistry is not recommended due to the probability of failed testing, especially in cases with a prolonged post-mortem interval and marked autolysis. The H&E slides may be used to evaluate the degree of autolysis and selected cases with better preserved tissue may proceed to testing. Further testing by MSI analysis or molecular genetic analysis would not be advised because of the potential poor preservation of tumour tissue, even in samples with a short post-mortem interval of 1 day. In suspected cases of CMMR-D syndrome, the alternative would be to clinically screen and counsel the parents for genetic testing to ensure good quality samples and reliable results. It may reveal more pertinent information on the risk and need for testing in any surviving siblings.
4.2. Staining Pattern Unlikely to Be Associated with CMMR-D Syndrome
A total of 12 samples showed strong, diffuse, intact nuclear staining in all four antibodies. These cases are unlikely to represent CMMR-D syndrome. A potential for a false negative result occurs in the setting of CMMR-D syndrome with underlying missense mutations which may show intact immunohistochemical stains. The frozen section sample (sample 15) formed part of this group and the staining was adequate. This gives an indication that frozen section samples may be suitable for testing by MMR immunohistochemistry; however, a bigger sample size would be needed to make a definitive recommendation.
4.3. Staining Pattern of Unknown Significance
A total of two samples showed loss of staining in a pattern that was not compatible with CMMR-D syndrome. One sample showed absent or reduced staining in PMS2 and MSH2 which are not heterodimer pairs. This sample was noted to have cautery artefact which may have impaired the staining process. One sample showed absent or reduced staining in MSH2, PMS2 and MSH6 which is also not described. The reduced staining in more than expected MMR stains may indicate a poorly preserved specimen.
4.4. Staining Pattern Compatible with CMMR-D syndrome
A total of four samples showed loss of staining in a pattern that is compatible with CMMR-D syndrome. Intermediate to strong nuclear staining was interpreted as intact nuclear staining. These cases will be referred to the Department of Human Genetics, for further confirmatory testing. These cases are outlined below.
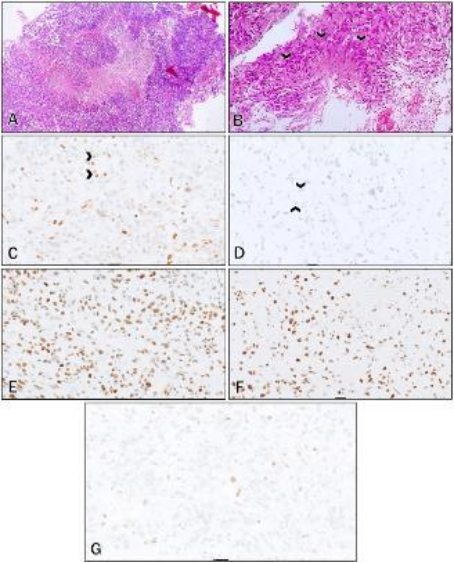
Figure 6. (A-G): MMR immunostains of Sample 1 (Brainstem glioblastoma).
A: H&E low power view –Malignant glial cells with increased nuclear-to-cytoplasmic ratios, nuclear pleomorphism and increased mitotic activity (arrows)
B: H&E low power view – areas of microvascular proliferation and glomeruloid structures (arrows)
C: H&E low power view – A focus of palisaded necrosis (arrow)
D: MLH1 – Intensity = 2, Proportion = 1; absent staining in the endothelial cells (arrow)
E: PMS2 – Intensity =1; Proportion = 1; absent staining in the endothelial cells (arrows)
F: MSH2 – Intensity = 2; Proportion = 5
G: MSH6 – Intensity = 2; Proportion = 4
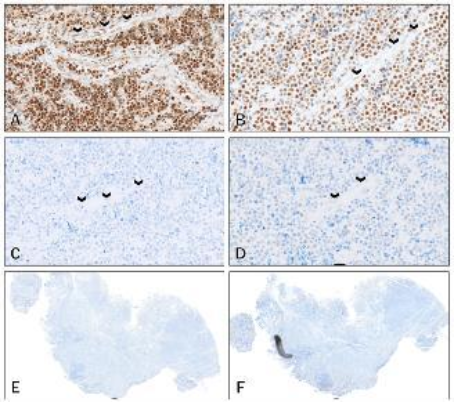
Sample 1 (See
Figure 6) showed loss of PMS2 and MLH1, a heterodimer pair. This sample was a post-mortem sample of a brainstem glioma. MSH2 and MSH6 were expected to show strong intact nuclear staining; however, weaker and patchy staining of the tumour cells was present. This inconsistency in staining is possibly as a result of a prolonged post-mortem interval.
Sample 18 (See
Figure 7) and sample 8 (See
Figure 8) were taken from the same patient with the same diagnosis of Burkitt lymphoma. Sample 18 was the first biopsy and sample 8 was a second biopsy taken after a 6-month interval. Both samples showed absent staining of MSH2 in the tumour cells with absent staining in the background endothelial cells. MSH6, the heterodimer of MSH2, shows markedly reduced staining in the tumour cells and absent staining in the background endothelial cells.
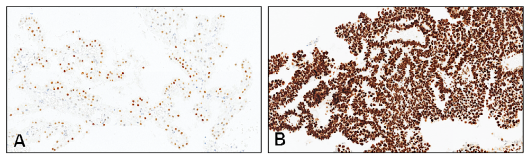
Figure 7. (A-F) MMR immunostains of Sample 18 (First biopsy; Burkitt Lymphoma).
A: MLH1 – Intensity = 3; Proportion = 5; positive endothelial cells (arrows)
B: PMS2 – Intensity = 3; Proportion = 5; positive endothelial cells (arrows)
C: MSH2 – Intensity = 0; Proportion = 0; negative endothelial cells (arrows)
D: MLH6 – Intensity = 1; Proportion = 4; negative endothelial cells (arrows)
E: MSH2 – Low power view showing absent staining of the tumour cells
F: MSH6 – Low power view showing markedly reduced staining of the tumour cells
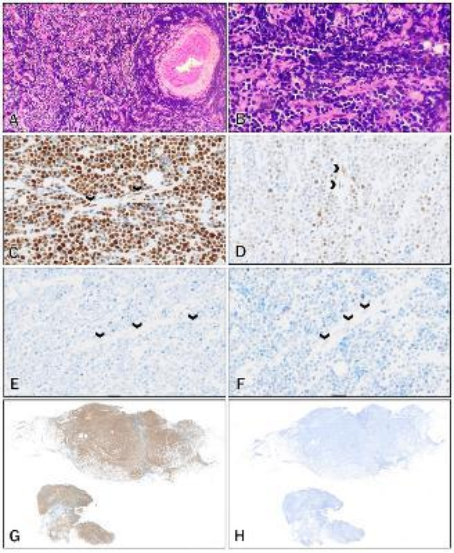
Figure 8. (A-H): MMR immunostains of Sample 8 (Second biopsy after a 6-month interval; Burkitt Lymphoma).
A: H&E low power view – Cervical biopsy showing infiltration by discohesive malignant cells
B: H&E high power view – The discohesive malignant cells show increased nuclear-to-cytoplasmic ratios, nuclear hyperchromasia, features consistent with a high-grade lymphoma
C: MLH1 – Intensity = 3; Proportion = 5, positive endothelial cells (arrows)
D: PMS2 – Intensity = 2; Proportion = 5, positive endothelial cells (arrows)
E: MSH2 – Intensity = 0; Proportion = 0, negative endothelial cells (arrows)
F: MSH6 – Intensity = 1; Proportion = 2, negative endothelial cells (arrows)
G: MLH1 – Low power view showing positive staining of the tumour cells
H: MSH2 – Low power view showing absent staining of the tumour cells
Sample 16 (See
Figure 9) showed loss of expression of PMS2, which is the most common mutation seen in CMMR-D syndrome. This tumour was a glioblastoma of the frontal lobe. The background neurons, intratumoural lymphocytes and endothelial cells showed positive nuclear staining, which is contrary to the described loss of staining in the normal tissue seen in CMMR-D syndrome. This tumour was initially scored with a high proportion of 3, but when the case was reviewed, the nuclear positive cells were identified as tumour infiltrating lymphocytes and endothelial cells. This is a potential pitfall in scoring of MMR immunohistochemistry, an overestimation of the proportion of tumour cells by including background non-tumour cells. In this sample, this misinterpretation was easy to make because the tumour cell nuclei were similarly sized to lymphocytes and endothelial cells. It is important to take care in evaluating a DAPI stained slide because clues to cell type, such as morphology and character of cytoplasm, are not as easily assessable as it is on a Haematoxylin and Eosin-stained slide and the cells can be misidentified.
Figure 9. (A-G): MMR immunostains of Sample 16 (Frontal lobe glioblastoma).
A: H&E low power view: Tumour with increased cellularity and a central area of palisaded necrosis
B: H&E high power view: Malignant cells show increased nuclear-to-cytoplasmic ratio, nuclear pleomorphism and palisaded necrosis (arrows)
C: MLH1 – Intensity = 2; Proportion = 5; positive endothelial cells (arrows)
D: PMS2 – Intensity = 1; Proportion = 3; negative endothelial cells (arrows)
E: MSH2 – Intensity = 3; Proportion = 5
F: MSH6 – Intensity = 3; Proportion = 5
G: PMS2 – Higher power view showing focal positive tumour cells and positive intratumoural lymphocytes
Only sample 16 of the four samples showed isolated loss of PMS2 staining, a pattern associated with PMS2 gene mutations which is the classically described mutation seen in CMMR-D syndrome. This may reflect a reduced frequency of PMS2 mutation in the South African LS population.
4.5. Staining Characteristics of the Background Non-tumour Tissue
It is described that loss of MMR staining in the background tissue is a classical finding in CMMR-D syndrome; however, this has been poorly characterized in studies. This gives pathologists little guidance when evaluating a specimen for potential CMMR-D syndrome. Some biopsies may be small, cauterized, necrotic and contain minimal normal tissue which hampered this assessment. We were also evaluating slides stained with DAPI which also makes it difficult to identify cellular details and normal tissue types, for example lymphocytes look very similar to glial cells. This was not seen in the setting of LS because normal colonic mucosa is typically easy to identify. This problem could be overcome by comparison with the H&E slides or double staining techniques, but this is cumbersome and may still be unreliable.
In this study, the easiest tissue landmark to identify and evaluate were the blood vessels. One of the hallmarks of cancer is induced angiogenesis and even biopsies of solid tumours contain dispersed blood vessels. In samples with intact staining of MMR proteins, the endothelial cells showed positive nuclear staining which was easy to identify. Even in the samples with patchy staining, at least some of the endothelial cells showed some positive staining. This may be attributed to endothelial cells being more resistant to ischaemic damage or autolysis because they are in direct contact with oxygenated blood. This is an important finding because in brain biopsies, lymphocytes are difficult to distinguish from glial cells. Lymphocytes are not native to the central nervous system and may not be represented. Intravascular circulating lymphocytes may be sparse. In the samples with intact MMR staining, glial cells seem to stain negative or variably. In the samples with intact MMR staining, neurons (See
Figure 10) and choroid plexus and choroid plexus carcinoma (See
Figure 11) also stained consistently and strongly positive. Our sample size was small, and a larger sample size will be needed to ensure that this finding is consistent.
Figure 10. Positive nuclear staining in normal neurons and endothelial cells.
MLH1 of Sample 13 – Nuclear positivity in normal background neurons (arrow heads) and endothelial cells (>)
Figure 11. Positive nuclear staining in both normal choroid plexus and choroid plexus carcinoma.
A: MLH1 of Sample 4 – Background normal choroid plexus showing nuclear positivity
B: MSH2 of Sample 5 – Choroid plexus carcinoma showing strong nuclear positivity
In the lymphomas, the staining pattern was more difficult to evaluate because normal lymph nodes show positive staining in follicles and reduced staining in the interfollicular zones. High-grade lymphoma tumour cells are easy to identify; however, this may be more difficult in indolent, small cell lymphomas. The tumour cells are small, and the distribution of neoplastic cells might be difficult to evaluate, particularly in small biopsies or core biopsies. This may result in the underrepresentation of small cell lymphomas in the CMMR-D syndrome tumour spectrum. Endothelial cells were still more easily identifiable landmarks. Until there is better characterization of MMR staining in the different kinds of lymphocytes, endothelial cells are a more reliable and consistent tissue type to evaluate for background staining.
4.6. Interpretation of the Results
Only three samples showed staining patterns consistent with CMMR-D syndrome including the loss of staining of the background tissue. The recommendation would be to confirm a biallelic mutation in one of the MMR genes by molecular genetic methods and these cases have been referred to the Department of Human Genetics for further evaluation. Once the cases of CMMR-D syndrome are confirmed, the Allred scores for these samples can be reviewed, and the cut off points for negative and positive cases can be proposed. We expected a predominance in MLH1 loss in our population or even predominantly PMS2 loss which is classically described. However, we only had three patients with staining compatible with CMMR-D syndrome: one with MLH1/PMS2 loss, one with MSH2/MHS6 loss and one with isolated PMS2 loss.
16 of the 21 samples represent high-grade gliomas which is like the Wimmer et al. study where a large proportion of tumours were high grade gliomas. Of the 16 high-grade gliomas, only two showed a pattern of loss or reduced MMR staining that was compatible with CMMR-D syndrome, far fewer than the study in Jordan that showed 51% of paediatric gliomas associated with germline mutations in MMR genes. This highlights a difference in frequency of MMR deficient high-grade gliomas in a country with predominantly founder effect compared to a country with high rates of consanguinity. The high-grade gliomas in our population are predominantly MMR stable and it may be interesting to test the tumours for other mutations common in the paediatric age group such as the diffuse midline glioma, H3 K27M mutant.
Even though CMMR-D syndrome is a polyposis syndrome, there is no representation of colorectal polyps or colorectal carcinomas. This was attributed to selection bias. The patients attending Red Cross Children’s hospital are younger because the hospital admissions policy has a cut-off age of 14 years. The colorectal manifestations of CMMR-D syndrome occur in adolescence or early childhood. This could be addressed by including samples from the adult referral laboratories.
Lymphomas were severely underrepresented in this study. In Wimmer et al.’s study, lymphomas were the second most represented tumours. The majority were Non-Hodgkin T-cell lymphomas; however Non-Hodgkin B-cell lymphomas were also described. Non-Hodgkin T-cell lymphomas are generally less common compared to Non-Hodgkin B-cell lymphomas. In the articles reviewed, some did not classify the lymphomas into specific histological diagnoses, such as Burkitt lymphoma or diffuse large B cell lymphoma. This may have hampered our computerised search for cases because there may not be specific codes for Non-Hodgkin T- cell lymphoma. The classification of lymphomas has also expanded and many of the cases may require reclassification by present day criteria. Immunohistochemistry may have been available internationally at the time but may not have been readily available in our institution, particularly in the 1980s. Even with available immunohistochemistry, there may still be interobserver variability and confirmation by molecular techniques may still be required. In future studies, the classification and spectrum of lymphomas associated with CMMR-D syndrome should be defined. A reason for selection bias is that in our hospital, bone marrow aspirates and trephines are evaluated by the Department of Haematology. This may exclude potential cases that were diagnosed on bone marrow biopsies. Another variable is that the phenotype of CMMR-D syndrome arising from the unique MLH1 c1528C>T mutation is not well characterized. There may be less expression of lymphomas in this phenotype and that may explain why despite clinical screening of patients and the computerized search of cases, only a single lymphoma was represented. Our study only analysed archival FFPE tissue and this would exclude any presentation of leukaemia which are typically diagnosed on peripheral blood samples.
Multiple metachronous tumours are described in CMMR-D syndrome, but only one patient had a second interval biopsy during the study interval. Both samples were taken from the left neck region 6 months apart and were reported as Burkitt lymphoma. The clinical interpretation was that the second biopsy represented a recurrence of the tumour. We also expected a more aggressive phenotype in our population due to the predominance of MLH1 mutations. Even though there was no outright predominance of MLH1 loss, all three of the cases with staining compatible with CMMR-D syndrome died shortly after diagnosis or discharged home for palliation. The patient from the South African case report of MLH1 mutated CMMR-D syndrome described in the introduction also did not survive very long after diagnosis. In our population, multiple metachronous tumours may not be a feature of CMMR-D syndrome because of an aggressive phenotype and patients not surviving long enough to develop a second tumour. There may be another unidentified reason for this.
During our selection process, we decided to exclude the non-malignant entities associated with CMMR-D syndrome. Future studies may be used to characterize the staining characteristics of these lesions, but due to their benign or indolent course, there is less urgency to do so.
4.7. Strengths and Limitations of the Study
This is the first study in our country to use the C4CMMRD criteria to identify at-risk patients for testing of MMR deficient tumours by immunohistochemistry. There was a focus on the evaluation, scoring and interpretation of the immunohistochemical staining patterns because it is key in distinguishing CMMR-D syndrome from LS. Some articles had inconsistencies and variable descriptions of the normal tissue evaluated for loss of staining
| [1] | Amayiri, N, U Tabori, B Campbell, D Bakry, M Aronson, C Durno, P Rakopoulos, et al. 2016. “High frequency of mismatch repair deficiency among pediatric high grade gliomas in Jordan.” International Journal of Cancer 138: 380-385. https://doi.org/10.1002/ijc.29724 |
[1]
. Some articles report reduced staining and some articles report absent staining as indicative of MMR gene mutation. By applying a modified Allred scoring system, the staining could be described in more precise and comparable measurements. We were able to differentiate cases that have complete absence of staining from cases that show reduced staining and describe, by proportion and intensity, the reduction in staining. Fortunately, the MMR proteins show nuclear staining which is easier to evaluate than cytoplasmic staining which could be confounded by aberrant staining.
This is also one of the only studies that focused on the characterization of the background staining in CMMR-D syndrome. And identified specific structures such as endothelial cells, neurons and choroid plexus that can be used to evaluate the presence of loss of background staining. This is critical information when the defining feature of CMMR-D syndrome is the loss of background staining.
This study also re-emphasizes the importance of establishing the adequacy of the tissue sample and staining technique in the assessment of MMR immunohistochemistry. Avoiding samples with poor preservation, the areas with cautery and necrosis and ensuring an adequate external positive control. The pitfall is to interpret the loss of staining in tumour and background tissue and a failed stain despite a positive external control. We also identified that post-mortem samples are generally not suitable for assessment by MMR IHC which was not recognized in other studies.
There was also identification of the gap in characterization of lymphomas associated with CMMR-D syndrome. Selection bias resulted in the underrepresentation of lymphomas in this study. Inclusion of bone marrow biopsies and peripheral blood samples may have improved the yield of haematolymphoid malignancies.
CMMR-D syndrome is a rare entity and our overall sample size was small which has an impact on the significance of the findings. Paediatric tissue samples submitted for tumour diagnostics in the Western Cape Province are split by drainage areas between Red Cross Children’s Hospital and Tygerberg Hospital, the major histopathology laboratories in the province. Other cases may be sent to the private laboratories in the region. A multi-centre study may be proposed to increase the number of cases and give a true reflection of the population in the Western Cape Province and also in the Northern Cape Province where the known LS families reside.
4.8. Implication of the Results and Recommendations
Pathologists may be familiar with the evaluation of MMR immunohistochemistry for screening of LS in colorectal carcinomas, but they also need to be aware of the different pattern and interpretation of staining in paediatric cases suspected of CMMR-D syndrome. They may even be able to detect cases of CMMR-D syndrome when performing routine screening for LS.
Causes for false negative and false positive results are recognized and additional molecular genetic testing is still recommended if there is a strong clinical suspicion. Molecular genetic testing should also be considered in cases with reduced but not absent staining and cases where the background cells do not show the classic loss of staining seen in CMMR-D syndrome. In these cases, molecular genetic testing could detect either LS or CMMR-D syndrome. We agree with the C4CMMRD document that both immunohistochemistry and molecular genetic testing should be used to confirm the diagnosis of CMMR-D syndrome and immunohistochemistry should be performed first to identify cases for molecular genetic testing.
In future studies of CMMR-D syndrome patients that involve MMR immunohistochemistry, it is important to characterize the staining pattern in both the tumour tissue and normal background tissue. This characterization should involve a scoring system, such as the Allred scoring system to document the intensity and proportion of staining to control for interobserver variability. One of the potential pitfalls recognized in sample 16 of this study is that the tumour-infiltrating, non-tumour lymphocytes may show nuclear positivity and falsely raise the score of proportion of positive tumour cells. This can easily occur when the tumour cell nuclei are small and are of similar size to normal lymphocyte nuclei on DAPI stained cells. The background tissue assessed should include endothelial cell staining because they are present in intestinal and extraintestinal tumours and are readily identified. Other background cells in the central nervous system could include neurons and choroid plexus. Testing of post-mortem samples with prolonged post-mortem intervals is not recommended. Testing of frozen section tissue was adequate, but we only had one frozen section sample and testing of a larger sample size is required before a recommendation can be made. Studies of confirmed CMMR-D syndrome cases will be needed to determine the sensitivity and specificity of MMR immunohistochemistry, but in the meantime, immunohistochemistry should be used in conjunction with molecular genetic testing.
Studies involving CMMR-D syndrome-associated lymphomas should report the histological classification of lymphomas. To study the scope of CMMR-D spectrum of tumours, a prolonged study interval or older cohort is needed to include the gastrointestinal carcinomas. Collaboration with haematologists and the inclusion of haematological specimens is needed to include leukaemias.
It may be useful to review the University of Cape Town (UCT) Human Genetics LS registry for family members where both parents carry mutations in the same MMR gene. Their risk of having a child with CMMR-D syndrome is 25%. If enough cases can be recruited, this could be used to determine if the incidence, phenotype and severity is different due to the unique MLH1 gene mutation that is present in our LS population. This may also require more intense cancer screening protocols.
Discussion with our paediatric oncology colleagues will determine if routine MMR immunohistochemical screening of these tumours should be performed or if it should be performed on request. In the Red Cross Children’s hospital population, high-grade gliomas, non-Hodgkin T-cell lymphomas and supratentorial embryonal tumours do not occur frequently. In addition to screening children with carcinoma and multiple adenomas, it would be better to screen the abovementioned tumours in our paediatric population, even if they occur without the additional features in
Indications for CMMR-D screening in cancer patients outlined by Wimmer
et al. (See
Table 1). The motivation for this is that there are serious implications for the index patient and their families, and we should use every opportunity to screen for CMMR-D syndrome. Clinicians need to be aware that the parents typically do not meet the criteria for LS and multiple metachronous tumours may not be a feature of CMMR-D syndrome in our population. This emphasizes the importance of using a genogram or pedigree in children with suspected CMMR-D syndrome and suspected tumour syndromes in general.
5. Conclusion
CMMR-D syndrome is a rare and potentially lethal childhood tumour predisposition syndrome. It is probably underrecognized and this can be improved by increased clinician awareness around the entity with application of the criteria for indication of CMMR-D testing to identify potential cases. It also requires increased awareness by pathologists of the unique staining pattern of the MMR immunohistochemistry and confirmation by molecular genetic techniques. Diagnosis is critical to provide genetic counselling, testing and applying tumour screening protocols for affected patients and their families.
Abbreviations
CMMR-D | Constitutional Mismatch Repair Deficiency |
European C4CMMRD | European Consortium’s Care for CMMRD |
FAP | Familial Adenomatous Polyposis |
FFPE | Formalin Fixed Paraffin Embedded |
H&E | Haematoxylin and Eosin |
HNPCC | Hereditary Nonpolyposis Colorectal Cancer |
IHC | Immunohistochemistry |
LS | Lynch Syndrome |
MMR | Mismatch Repair |
MSI | Microsatellite Instability |
NF1 | Neurofibromatosis Type 1 |
PCR | Polymerase Chain Reaction |
UCT | University of Cape Town |
WHO | World Health Organization |
Acknowledgments
Prof A Davidson and the team and Red Cross Oncology unit for allowing access for review of the clinical folders.
Dr B Price for his friendship, mentorship and advice
Ms Xolelwa Mbutho and the team at the National Health Laboratories, Red Cross Children’s Hospital, Histology unit for archive retrieval and processing of tissue blocks.
Author Contributions
Sindy Jen-Yi Tu: Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Writing – original draft
Annesu Wessels: Conceptualization, Investigation, Methodology, Project administration
Raj Ramesar: Conceptualization, Supervision
Alan Davidson: Conceptualization, Investigation, Resources, Supervision
Komala Pillay: Conceptualization, Funding acquisition, Project administration, Methodology, Resources, Validation, Writing – review & editing
Funding
This work is supported by a Development Grant from the NHLS Research Trust. No additional funding was required.
Data Availability Statement
The data supporting the outcome of this research work has been reported in this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix
Equipment
Ventana Autostainer
Leica Rotary Microtome RM2125 RTS
Waterbath
Hotplate
Consumables and reagents
Glass slides – Histobond charged coated
Glass coverslips 22x40
Microtome Blades Low profile
Gloves – Latex free
Entellan
Xylene
Absolute Alcohol
Dishwashing liquid
Kits and automated reagents
Optiview DAB detection system
Haematoxylin II
Bluing reagent
EZ Prep
Liquid Coverslip (LCS)
CC1
Reaction Buffer
Antibodies
VENTANA anti-MSH2 (G219-1129) Mouse Monoclonal Primary Antibody
Clone Name: G219-1129
Species: Mouse
Localization: Nuclear
Dilution: Used as is
VENTANA anti-MLH1 (M1) Mouse Monoclonal Primary Antibody
Clone Name: M1
Species: Mouse
Localization: Nuclear
Dilution: Used as is
VENTANA anti-PMS2 (A16-4) Mouse Monoclonal Primary Antibody
Clone Name: A16-4
Species: Mouse
Localization: Nuclear
Dilution: Used as is
VENTANA anti-MSH6 (SP93) Rabbit Monoclonal Primary Antibody
Clone Name: SP93
Species: Rabbit
Localization: Nuclear
Dilution: Used as is
Reagents and Buffers
Immunohistochemistry – semi-automated method on Ventana XT auto-stainer reagents and buffers.
10X EZ Prep solution
EZ prep solution was used for paraffin removal in both IHC and ISH automated staining procedures. Two litres of EZ Prep reagent concentrate were made up to 20 litres using distilled water in the manufacture supplied plastic container. This was stored on the shelf and decanted into the machine bulk container as required.
Reaction buffer concentrate
Reaction buffer is a Tris based buffer solution which was used to rinse between IHC and ISH staining procedures. 2 L of Reaction buffer reagent concentrate was made up to 20 litres using distilled water in the manufacture supplied plastic container. This was stored on the shelf and decanted into the machine bulk container as required.
LCS
Also known as liquid coverslip, this solution is used as is, and is available in a 2 L plastic bottle, prediluted. LCS is a high temperature coverslip solution which is used to prevent the aqueous solution from evaporating and the section drying out. This allows for a stable IHC environment.
CC1
Also known as cell conditioning solution 1, this solution is used as is and is available in a 2 L plastic bottle. CC1 is a tris-based pre-treatment or antigen retrieval buffer used for automated immunohistochemical reactions.
OptiView DAB IHC Detection Kit
This kit was used for the semi-automated IHC staining. It contained propriety reagents in six 25 ml dispensers, which were automatically applied by the analyser. These reagents were peroxidase, universal linker, HRP multimer, hydrogen peroxide (H2O2), 3,3′-Diaminobenzidine (DAB) and copper (copper sulphate).
Hematoxylin II
Hematoxylin II counterstain reagent is a modified Mayer's hematoxylin and was available in a 25 ml dispenser. This reagent was used as is.
Bluing reagent
Bluing Reagent is an aqueous solution of buffered lithium carbonate and was available in a 25 ml dispenser. This reagent was used as is.
Dishwashing detergent (LCS wash)
Three drops of Sunlight® dishwashing soap was added to 500 ml of water
Absolute Alcohol This reagent was used as is
Xylene This reagent was used as is
Entellan – mounting media This reagent was used as is
References
| [1] |
Amayiri, N, U Tabori, B Campbell, D Bakry, M Aronson, C Durno, P Rakopoulos, et al. 2016. “High frequency of mismatch repair deficiency among pediatric high grade gliomas in Jordan.” International Journal of Cancer 138: 380-385.
https://doi.org/10.1002/ijc.29724
|
| [2] |
Bartley, AN, SR Hamilton, R Alsabeh, EP Ambinder, M Berman, E Collins, PL Fitzgibbons, et al. 2014. “Template for Reporting Results of Biomarker Testing of Specimens From Patients with Carcinoma of the Colon and Rectum.” December. Accessed June 2019.
|
| [3] |
Bruwer, Z, U Algar, A Vorster, K Fieggen, A Davidson, P Goldberg, H Wainwright, and R Ramesar. 2014. “Predictive genetic testing in children: Constitutional mismatch repair deficiency cancer predisposing syndrome.” Journal of Genetic Counselling 23: 147-155.
https://doi.org/10.1007/s10897-013-9659-2
|
| [4] |
Carrato C, C Sanz, AM Muñoz-Mármol, I Blanco, M Pineda, J Del Valle, E Dámaso, M Esteller, E Musulen. 2021. The Challenge of Diagnosing Constitutional Mismatch Repair Deficiency Syndrome in Brain Malignancies from Young Individuals. The Journal of Molecular Sciences. 22(9): 4629.
https://doi.org/10.3390/ijms22094629
|
| [5] |
Colas, C, L Guerrini-Rousseau, M Suerink, R Gallon, CP Kratz, É Ayuso, ERN GENTURIS CMMRD Guideline Group, L Brugières, K Wimmer. 2024. “ERN GENTURIS guidelines on constitutional mismatch repair deficiency diagnosis, genetic counselling, surveillance, quality of life, and clinical management”. European Journal of Human Genetics. 32(12): 526–1541.
https://doi.org/10.1038/s41431-024-01708-6
|
| [6] |
Durno, C, CR Boland, S Cohen, JA Dominitz, FM Giardiello, DA Johnson, T Kaltenbach, et al. 2017. “Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer.” Gastroenterology 152: 1605-1614.
https://doi.org/10.1053/j.gastro.2017.02.011
|
| [7] |
Durno C, AB Ercan, V Bianchi, M Edwards, M Aronson, M Galati, EG Atenafu, On Behalf of the International Replication Repair Deficiency Consortium. 2021. Survival Benefit for Individuals With Constitutional Mismatch Repair Deficiency Undergoing Surveillance. Journal of Clinical Oncology. Volume 39, Number 25.
https://doi.org/10.1200/JCO.20.02636
|
| [8] |
Fedchenko, N, and J Reifenrath. 2014. “Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue - a review.” Diagnostic Pathology (9): 221.
https://doi.org/10.1186/s13000-014-0221-9
|
| [9] |
Huang, S, C Durno, and SH Erdman. 2014. “Lynch Syndrome: A Pediatric Perspective.” Journal of Pediatric Gastroenterology and Nutrition 58: 144-152.
https://doi.org/10.1097/MPG.0000000000000179
|
| [10] |
Kang, E, JK Suh and SD Kim. 2025. “Constitutional Mismatch Repair Deficiency, the Most Aggressive Cancer Predisposition Syndrome: Clinical Presentation, Surveillance, and Management”. Journal of Korean Neurosurgical Society 68(3): 294–304.
https://doi.org/10.3340/jkns.2025.0024
|
| [11] |
Lamola, L. 2018. Genomics of Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome. Accessed April 10, 2020.
https://open.uct.ac.za/handle/11427/27840
|
| [12] |
Peltomaki, P. 2003. “Role of DNA mismatch repair defects in the pathogenesis of human cancer.” Journal of Clinical Oncology 21: 1174-1179.
https://doi.org/10.1200/JCO.2003.04.060
|
| [13] |
Ramchander, NC, NAJ Ryan, EJ Crosbie, and DG Evans. 2017. “Homozygous germ-linemutation of the PMS2 mismatch repair gene: a unique case report of constitutional mismatch repair deficiency (CMMR-D).” BMC Medical Genetics. 18(40).
https://doi.org/10.1186/s12881-017-0391-x
|
| [14] |
Tabori, U, JR Hansford, MI Achatz, CP Kratz, SE Plon, T Frebourg, and L Brugieres. 2017. “Clinical management and tumour surveillance recommendations of inherited mismatch repair deficiency in childhood.” Clinical Cancer Research 23: 32-37.
https://doi.org/10.1158/1078-0432.CCR-17-0574
|
| [15] |
Wimmer, K, CP Kratz, and HFA Vasen. 2014. “Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium 'Care for CMMR-D'.” Journal of Medical Genetics (Journal of Medical Genetics) 51: 355-365.
https://doi.org/10.1136/jmedgenet-2014-102284
|
Cite This Article
-
APA Style
Tu, S. J., Wessels, A., Ramesar, R., Davidson, A., Pillay, K. (2025). Identifying Children with Constitutional Mismatch Repair Deficiency Syndrome in the Expanding Lynch Syndrome Population in Cape Town, South Africa. Journal of Cancer Treatment and Research, 13(4), 119-139. https://doi.org/10.11648/j.jctr.20251304.14
 Copy
|
Copy
|
 Download
Download
ACS Style
Tu, S. J.; Wessels, A.; Ramesar, R.; Davidson, A.; Pillay, K. Identifying Children with Constitutional Mismatch Repair Deficiency Syndrome in the Expanding Lynch Syndrome Population in Cape Town, South Africa. J. Cancer Treat. Res. 2025, 13(4), 119-139. doi: 10.11648/j.jctr.20251304.14
Copy
|
Download
AMA Style
Tu SJ, Wessels A, Ramesar R, Davidson A, Pillay K. Identifying Children with Constitutional Mismatch Repair Deficiency Syndrome in the Expanding Lynch Syndrome Population in Cape Town, South Africa. J Cancer Treat Res. 2025;13(4):119-139. doi: 10.11648/j.jctr.20251304.14
Copy
|
Download
-
@article{10.11648/j.jctr.20251304.14,
author = {Sindy Jen-Yi Tu and Annesu Wessels and Raj Ramesar and Alan Davidson and Komala Pillay},
title = {Identifying Children with Constitutional Mismatch Repair Deficiency Syndrome in the Expanding Lynch Syndrome Population in Cape Town, South Africa
},
journal = {Journal of Cancer Treatment and Research},
volume = {13},
number = {4},
pages = {119-139},
doi = {10.11648/j.jctr.20251304.14},
url = {https://doi.org/10.11648/j.jctr.20251304.14},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.jctr.20251304.14},
abstract = {Introduction: Constitutional Mismatch Repair Deficiency (CMMR-D) syndrome is a rare tumour predisposition and polyposis syndrome that presents in childhood. It is caused by mutations in mismatch repair (MMR) genes that result in a tumour spectrum including colorectal cancers, high-grade gliomas, non-Hodgkin T-cell lymphomas and leukaemias. It is characterized by biallelic germline mutation of one of four MMR genes that can be identified by immunohistochemistry. Immunohistochemistry is used in screening for Lynch syndrome (LS); however, the pattern of loss-of-staining in the background, non-tumour tissue is unique to CMMR-D syndrome. CMMR-D syndrome is seen in LS families and occurs as a result of consanguinity or founder effect. In South Africa, LS families in the Western and Northern Cape Provinces show a unique MLH1 c1528C>T mutation. The diagnosis of CMMR-D syndrome includes clinical findings outlined in the European Consortium’s Care of CMMRD document and confirmation of biallelic mutation in one MMR gene. Immunohistochemistry can be used in the diagnosis of CMMR-D syndrome by identifying cases for targeted molecular genetic tests. Loss of staining of the affected gene in the background, non-tumour tissue, is a key feature of CMMR-D syndrome. Methods: A retrospective analysis of archival, formalin fixed paraffin embedded tissue was performed on specimens of children attending Red Cross Children’s Hospital with tumours that form part of the CMMR-D spectrum, outlined by the Care for CMMRD criteria. We used the criteria of high-grade gliomas (WHO Grade III or IV) occurring before 25 years of age, cutaneous lesions suggestive of CMMR-D syndrome and patients with a first or second degree relative diagnosed with LS. Immunohistochemistry was performed and the staining pattern was documented using a modified Allred Scoring system. Specific attention was given to the characterization of the staining pattern of the background normal tissue. Results: 21 samples evaluated from 18 patients. 16 samples represented brain tumours. Three inadequate samples were excluded. 12 samples showed intact staining. Two samples showed staining of unknown significance. Four samples from 3 different patients showed staining patterns compatible with MMR deficiency. Of these four samples, three samples showed loss of staining in background non-tumour tissue with positive external control. Conclusion: MMR immunohistochemistry can be used in the evaluation of CMMR-D syndrome. The pattern and scoring of both the tumour and the background non-tumour tissue is critical. The diagnosis of CMMR-D syndrome depends on clinical application of Care for CMMRD criteria, MMR immunohistochemistry in conjunction with molecular genetic testing.
},
year = {2025}
}
Copy
|
Download
-
TY - JOUR
T1 - Identifying Children with Constitutional Mismatch Repair Deficiency Syndrome in the Expanding Lynch Syndrome Population in Cape Town, South Africa
AU - Sindy Jen-Yi Tu
AU - Annesu Wessels
AU - Raj Ramesar
AU - Alan Davidson
AU - Komala Pillay
Y1 - 2025/11/07
PY - 2025
N1 - https://doi.org/10.11648/j.jctr.20251304.14
DO - 10.11648/j.jctr.20251304.14
T2 - Journal of Cancer Treatment and Research
JF - Journal of Cancer Treatment and Research
JO - Journal of Cancer Treatment and Research
SP - 119
EP - 139
PB - Science Publishing Group
SN - 2376-7790
UR - https://doi.org/10.11648/j.jctr.20251304.14
AB - Introduction: Constitutional Mismatch Repair Deficiency (CMMR-D) syndrome is a rare tumour predisposition and polyposis syndrome that presents in childhood. It is caused by mutations in mismatch repair (MMR) genes that result in a tumour spectrum including colorectal cancers, high-grade gliomas, non-Hodgkin T-cell lymphomas and leukaemias. It is characterized by biallelic germline mutation of one of four MMR genes that can be identified by immunohistochemistry. Immunohistochemistry is used in screening for Lynch syndrome (LS); however, the pattern of loss-of-staining in the background, non-tumour tissue is unique to CMMR-D syndrome. CMMR-D syndrome is seen in LS families and occurs as a result of consanguinity or founder effect. In South Africa, LS families in the Western and Northern Cape Provinces show a unique MLH1 c1528C>T mutation. The diagnosis of CMMR-D syndrome includes clinical findings outlined in the European Consortium’s Care of CMMRD document and confirmation of biallelic mutation in one MMR gene. Immunohistochemistry can be used in the diagnosis of CMMR-D syndrome by identifying cases for targeted molecular genetic tests. Loss of staining of the affected gene in the background, non-tumour tissue, is a key feature of CMMR-D syndrome. Methods: A retrospective analysis of archival, formalin fixed paraffin embedded tissue was performed on specimens of children attending Red Cross Children’s Hospital with tumours that form part of the CMMR-D spectrum, outlined by the Care for CMMRD criteria. We used the criteria of high-grade gliomas (WHO Grade III or IV) occurring before 25 years of age, cutaneous lesions suggestive of CMMR-D syndrome and patients with a first or second degree relative diagnosed with LS. Immunohistochemistry was performed and the staining pattern was documented using a modified Allred Scoring system. Specific attention was given to the characterization of the staining pattern of the background normal tissue. Results: 21 samples evaluated from 18 patients. 16 samples represented brain tumours. Three inadequate samples were excluded. 12 samples showed intact staining. Two samples showed staining of unknown significance. Four samples from 3 different patients showed staining patterns compatible with MMR deficiency. Of these four samples, three samples showed loss of staining in background non-tumour tissue with positive external control. Conclusion: MMR immunohistochemistry can be used in the evaluation of CMMR-D syndrome. The pattern and scoring of both the tumour and the background non-tumour tissue is critical. The diagnosis of CMMR-D syndrome depends on clinical application of Care for CMMRD criteria, MMR immunohistochemistry in conjunction with molecular genetic testing.
VL - 13
IS - 4
ER -
Copy
|
Download